治験契約から終了までの流れ
エントリー開始までの流れ
CRCとの調整事項-
-
契約
治験薬搬入
手続きの流れ等
開始準備
(院内各部門調整・関連書類準備等)
-
スタートアップミーティング
同意取得
治験説明・同意取得の流れ 資料①適格性の確認
適格性確認の流れ 資料②治験薬投薬開始
治験薬の流れ(内服) 資料④、(注射) 資料⑤被験者対応
患者来院時の流れ 資料③
モニタリング/監査対応等
治験継続の意思確認の流れ 資料⑥
AE・SAE発生時の流れ 資料⑦
臨床検査の流れ 資料⑧
生理検査・画像検査の流れ 資料⑨
原資料作成~症例報告書作成までの流れ 資料⑩
逸脱発生時の流れ 資料⑪治験薬投与終了
治験中止・終了の流れ 資料⑫治験薬回収
治験終了
必須文書の保管について 資料⑬
医療機関内での品質管理について
治験データの記録に関するプロセスを確認するためのツールとして、日本製薬工業協会の資料を基に、「治験データの記録プロセス確認リスト」を作成しました。本リストを基に、治験データがどの原資料に記録されているか/記録されるかを治験開始前に明確にします。
治験の実施に関わる重要事項の記録の保管について
治験に関する重要事項の記録(運用通知 第41条)の当院での取扱いについて明確にするために、「治験の実施に関わる重要事項の記録の保管に関する院内規定」を作成しています。 本規定に基いて、治験開始前に治験依頼者の担当者と記録の保管の手順について協議します。
Delegation Log
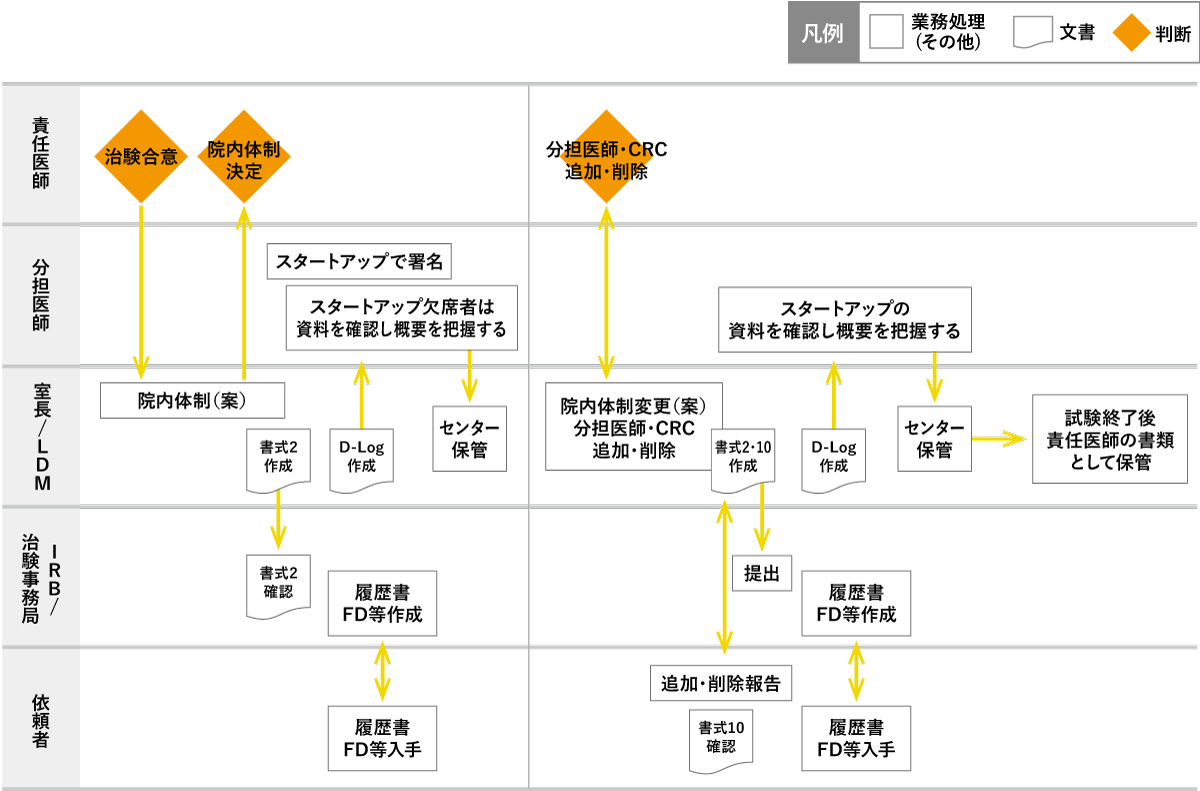
担当CRCはヒアリングまでに決定します。また、ヒアリング後に、責任医師と室長が担当者を決定し、治験分担医師・治験協力者リスト(書式2)を作成します。
Delegation Log は北大書式(TransCelerate 参考)を使用し、GCP 第43 条に則って作成・保管・管理について院内で行います。院内各部門(看護部、検査・輸血部、薬剤部、放射線部、病理部)の通常臨床業務はDelegation Log に明示しますので、分担医師・CRC 以外の院内スタッフについては原則各個人のDelegate は不要とします。
- Delegation Log_ Ver.1.2(2019年度契約までの試験)
- Delegation Log_ Ver.2.0(2020年度以降契約の試験)
- Delegation Log_ Ver.3.0(2021年9月IRB承認以降新規契約の試験)
- Delegation Log_ Ver.4.0(2025年度以降契約の試験)
- 体制構築の流れ
Training Log
Training Logは北大書式を使用し、GCP 第43 条に則って作成・保管・管理について院内で行います。院内スタートアップミーティング開催時、プロトコル及び治験薬管理SOP変更時(軽微な変更は除く)および治験スタッフが追加でDelegate された場合、院内各部門と対面での調整を行った場合に作成します。試験特有のWebトレーニング等については対応します。
資料①:治験説明・同意取得の流れ
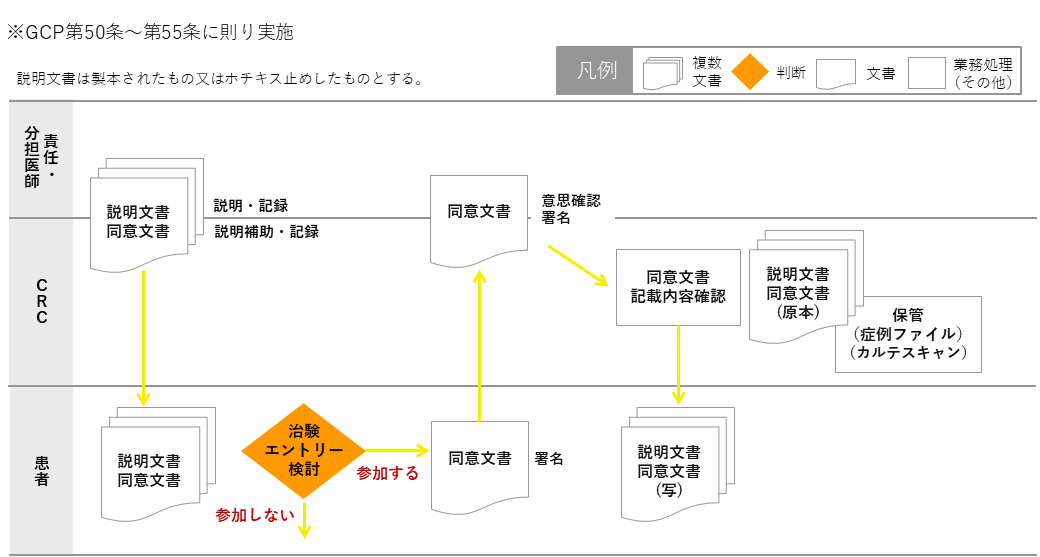
CRCは責任(分担)医師の治験説明にできるだけ同席し、責任(分担)医師の治験説明後、CRCは患者の理解や疑問を確認し説明補助を行う。患者自身が考え自由意思で参加、不参加を決定できるよう時間的な配慮を行う。また、参加後もいつでも中止できることを説明する。
候補者の治験参加への意思確認がとれたら、同意書に署名もらう。
CRCはDDworks Trial Site(DDTS)で最新版であること等をダブルチェックし、記載されている日時・署名や版数記載に不備がないか等を確認し、説明文書と同意書(写)を患者へ手渡す。責任(分担)医師はカルテに同意取得した旨を記載する。また、CRCは説明時に被験者からの質問事項や同意に至った経緯についてカルテに記載し、電子カルテにスキャンする。さらに、治験に参加している患者であることがシステム上で認識されるように、電子カルテに登録する。
資料②:適格性の確認の流れ(割付まで)
※GCP第44条に添って実施
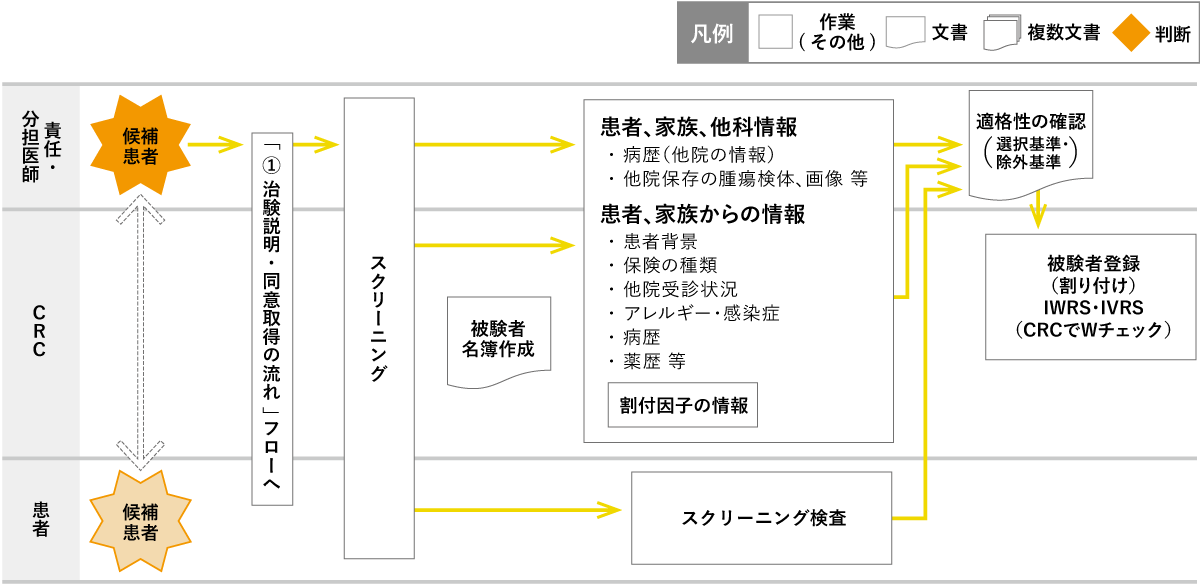
治験参加の選択基準を満たし、除外基準に該当しないかをカルテや被験者本人から聴取する。選択基準・除外基準のワークシートを用いて、CRCは適格性の確認補助を行い、最終確認は責任(分担)医師が行う。被験者登録時はCRC間で被験者名・識別コード・登録項目をダブルチェックし、割り付けを行う。ワークシート、割付結果の原本は症例ファイルで保管し、カルテにもスキャンする。
資料③:患者の来院時の流れ
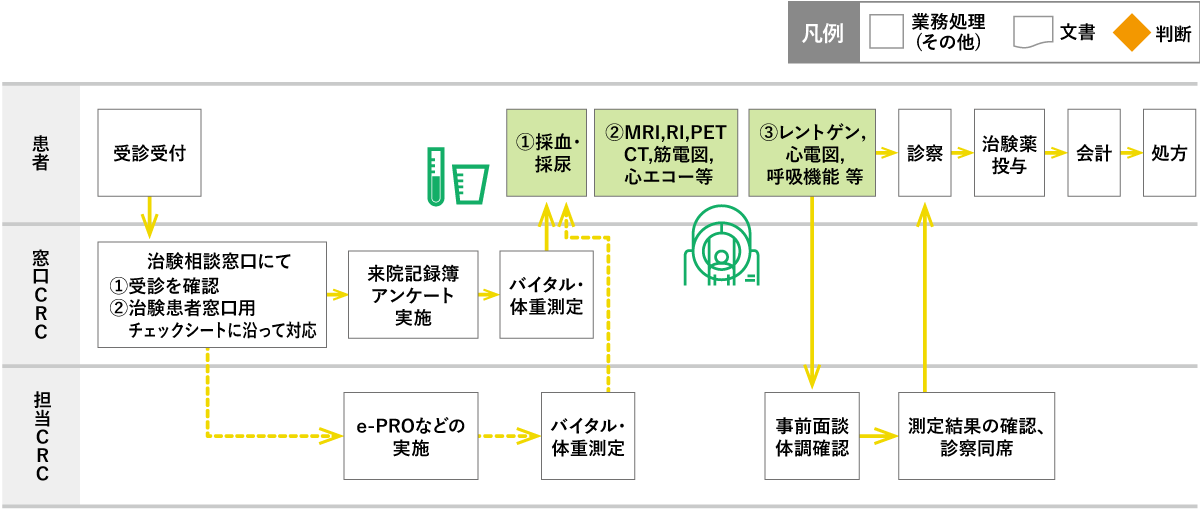
患者は受診受付(自動再来受付機)後、臨床研究・治験相談窓口に立ち寄る。
窓口CRCは来院記録簿にサインをもらい、チェックシートに沿って、アンケート、体重・バイタル測定を実施しカルテ/温度板に記録する。
e-PROなど担当CRCの対応が必要な場合は、治験外来等へご案内する。
通常、検査結果確認後の診察などを考慮し、患者は、以下の順番で検査を行う。
①採血・採尿、②時間指定の検査、③その他の検査
医師とCRCは検査結果等を確認し、医師が診察を行う。
投薬については、「治験薬の流れ③④」参照
会計後、処方薬を受け取り帰宅
資料④:治験薬の流れ(内服)
※GCP第35、39、41、45条に添って実施
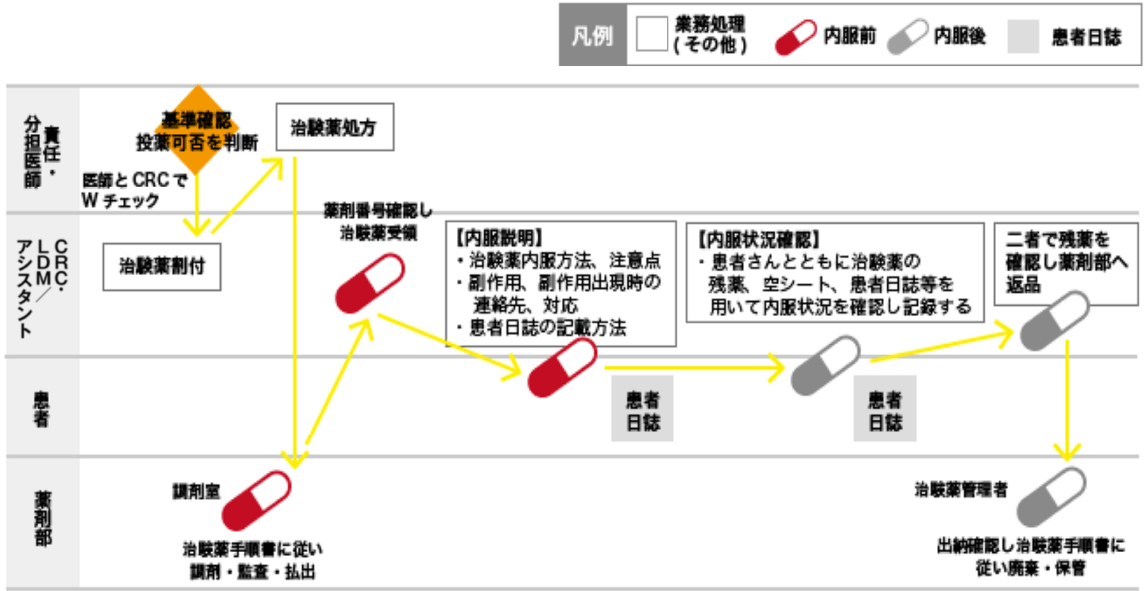
治験薬投与当日に割り付けシステム(IWRS、IVRS、FAX)で治験薬剤番号を入手し、責任(分担)医師は、治験薬登録確認書に基づいて治験薬をオーダーする。
CRCは、オーダー内容(治験薬名、薬剤番号、用法用量、日数等)をプロトコル、患者登録票を基に確認する。
CRCは患者日誌等を用いて、治験薬の残薬・空シートや内服状況を確認し記録する。
治験薬管理補助者は、処方と返却された薬剤のWチェックを行う。
資料⑤:治験薬の流れ(注射)
※GCP第35、39、41、45条に添って実施
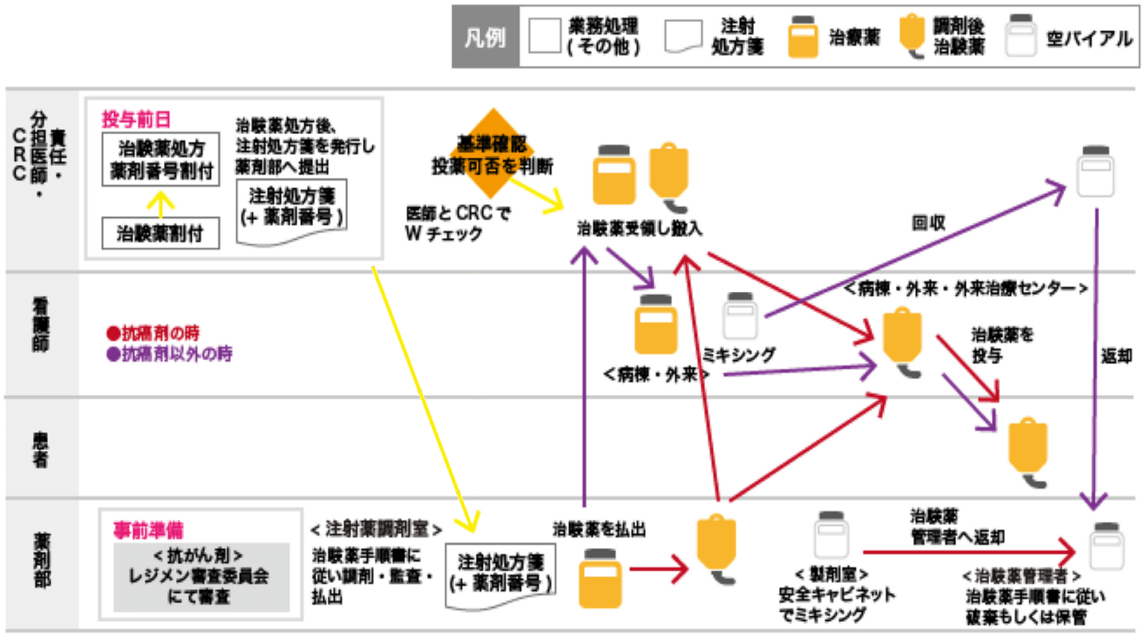
治験薬投与前日までに割り付けシステム(IWRS、IVRS、FAX)で治験薬剤番号を入手し、責任(分担)医師は、治験薬登録確認書に基づいて治験薬をオーダーする。
払出し伝票の「治験薬品用注射処方箋」に割付結果のコピーを添付し、注射薬調剤室へ提出する。
治験薬はCRCが受け取り、該当部署に搬入する。
【抗がん剤の場合】
事前に院内レジメン審査委員会にて、レジメンが審査され、承認されたレジメンはセットオーダーの登録が行われる。
資料⑥:治験継続の意思確認の流れ(再同意)
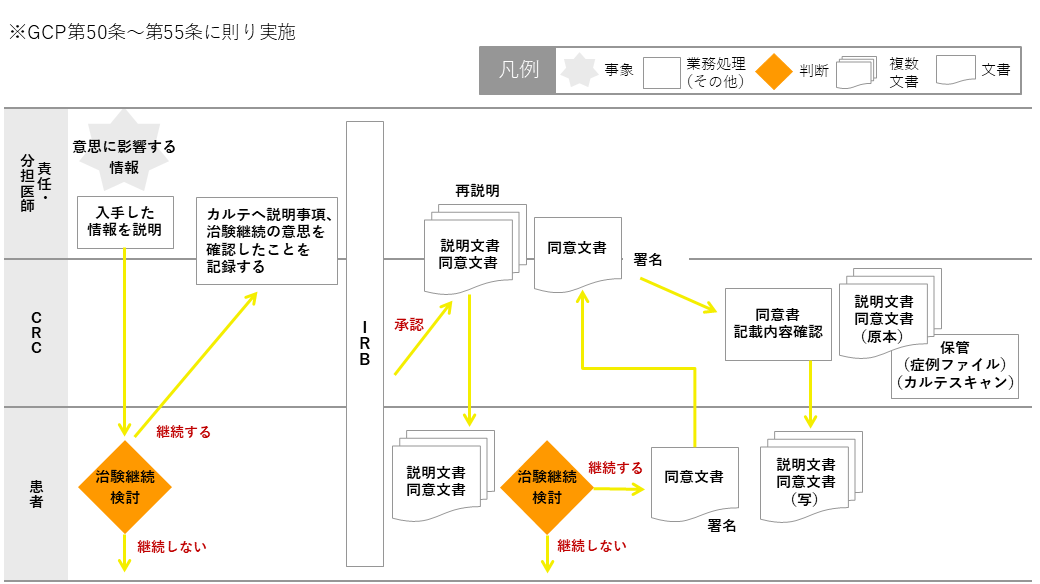
同意説明文書を変更すると責任医師が判断した場合、院内マニュアルに則り、該当する被験者へ同意説明文書の変更箇所を説明、治験継続の意思を確認し、その旨をカルテに記録する。
改訂された同意説明文書がIRBで承認後、文書にて同意を取得する。
資料⑦:AE・SAE発生時の流れ
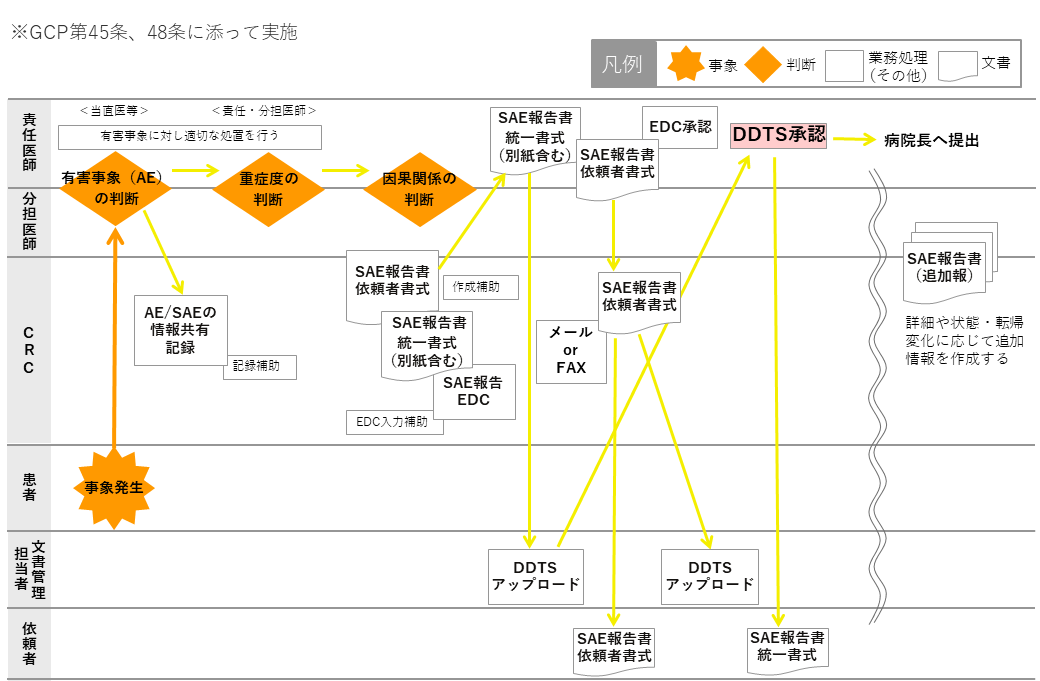
患者から普段と異なる症状等が発生した場合、平日はCRCが、夜間は該当の病棟経由当直医が連絡を受ける。責任(分担)医師に状況を報告し、有害事象かどうか重篤度、治験薬との因果関係の判断を行う。責任(分担)医師は有害事象に対し適切な処置を行う。SAEに該当する場合には、プロトコルに準じて速やかに依頼者へ報告する。
CRCはSAE報告書の作成補助を行い(統一書式・依頼者書式)、責任医師の確認後、病院長・依頼者へ提出する。
※SAE報告がEDCの場合は、SAE発生時までの情報がもれなく入力されていることを確認する。
詳細や状態・転帰の変化に応じて追加報の作成補助を行う。
資料⑧:臨床検査の流れ
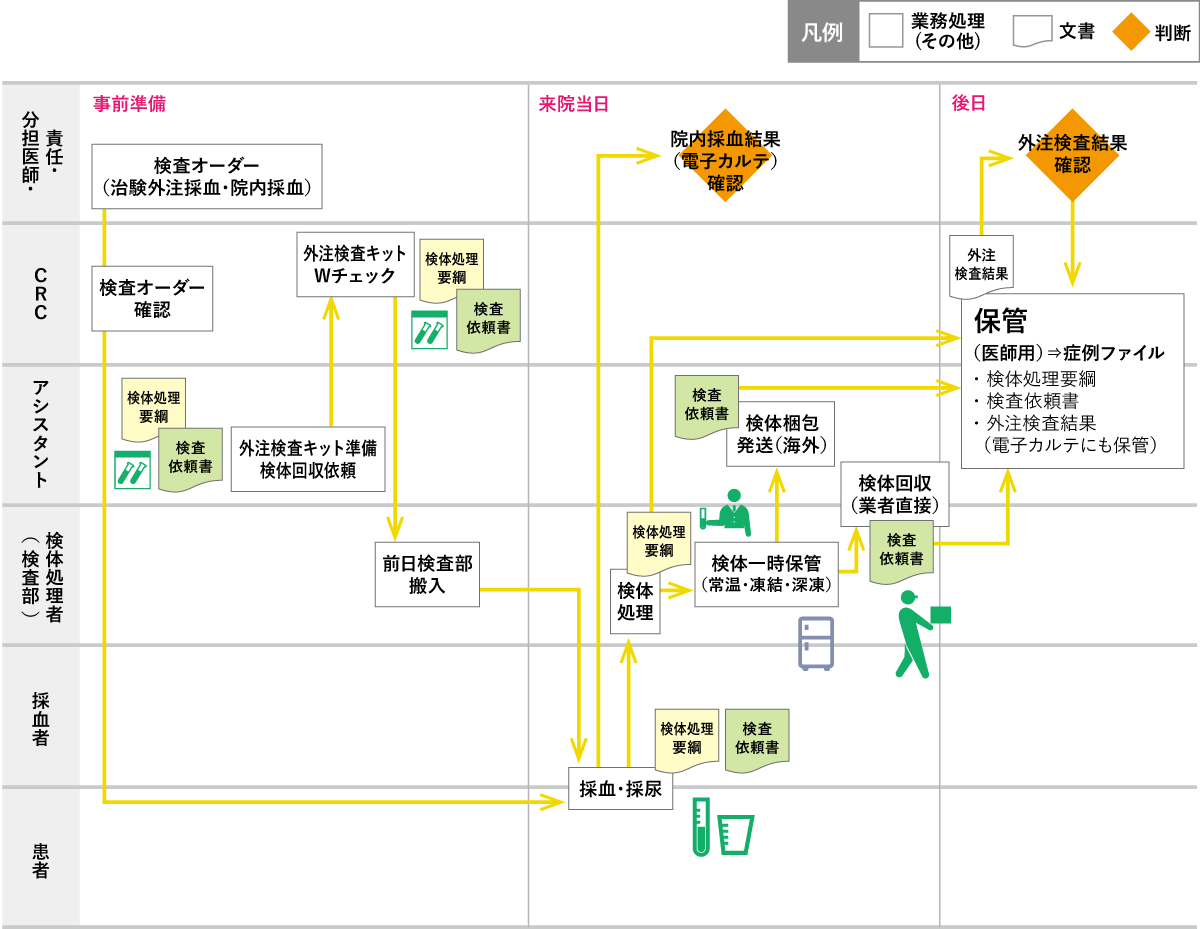
院内検査については、プロトコルで規定された検査項目を責任(分担)医師がオーダーし、CRCは検査前日までに、プロトコルとオーダーを照合し必要時CRC間でダブルチェックを行う。
外注検査がある場合は、責任(分担)医師が「治験採血」「治験尿」をオーダーする。
当該治験の回収依頼の日数を考慮し、Visitの前日までに検体を準備する。
作成者・確認者は別の者が行う。(担当CRCは、作成もしくは確認のどちらかには必ず関与し、プロトコルと照らし合わせて準備・確認を行う。)
採血後は処理要綱に従って検査技師が検体処理を行い、検体を保管する。
原則検体は当日発送・提出とする。(時間外に発生するもの、バックアップ等やむを得ない場合を除く)
海外へ発送する検体は担当者が発送本数・症例番号・検査伝票に記載漏れがないかを確認し、運送業者に渡す。
検査結果については責任(分担)医師が確認し、適切に保管する。
CRCおよびアシスタントは、外注検査キットの管理(検査会社からのキット受領、不足資材の発注、期限切れや終了時の廃棄等)を行う。
資料⑨:生理検査・画像検査の流れ
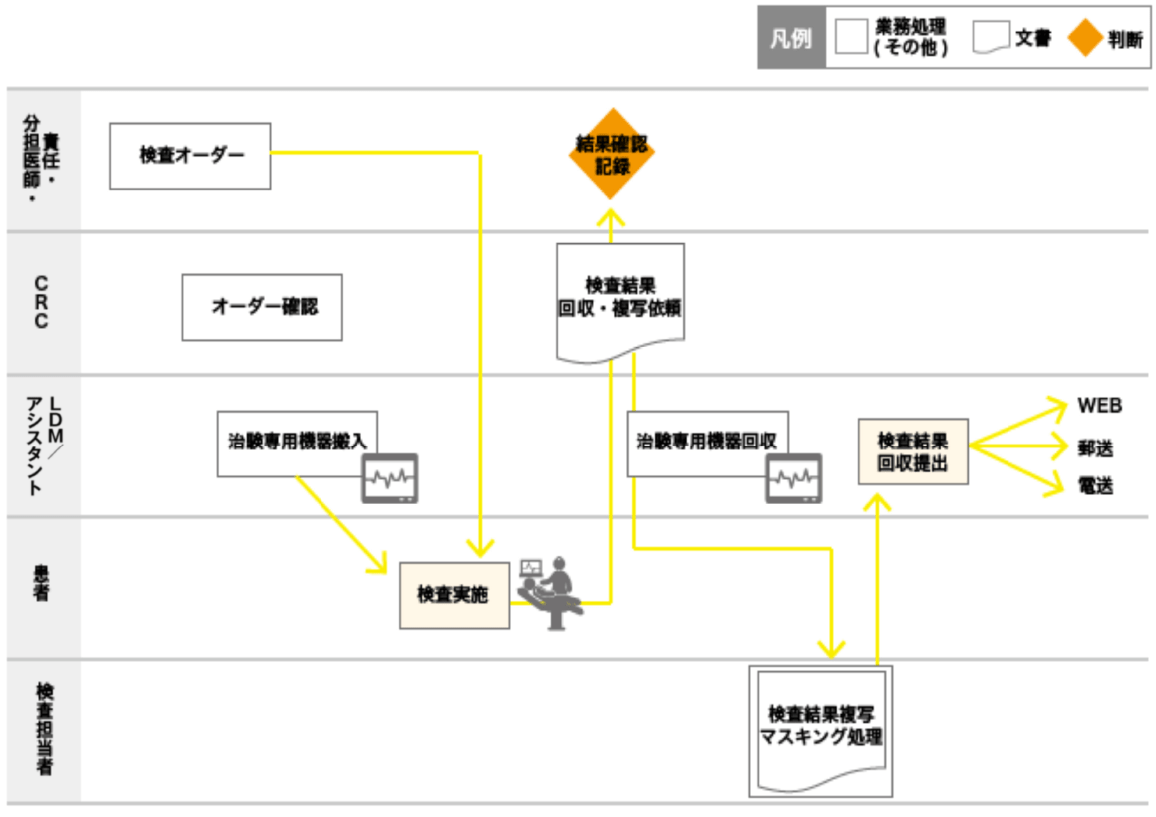
事前に各部門担当者と測定条件・必要事項コメントを取り決め、オーダーの際、責任(分担)医師に必要事項の入力を依頼する。 専用の搬入機器を使用する場合には、前日に検査室へ搬入する。 検査後は責任(分担)医師が結果を確認/記録を行い、試験毎に規定された方法で結果を提出する(web・郵送・電送)。 心電図の場合、検査開始前に5分安静の後、波形が安定したことを確認し測定を開始している。
資料⑩:原資料作成~症例報告書作成までの流れ
※GCP第37、41、47条に添って実施
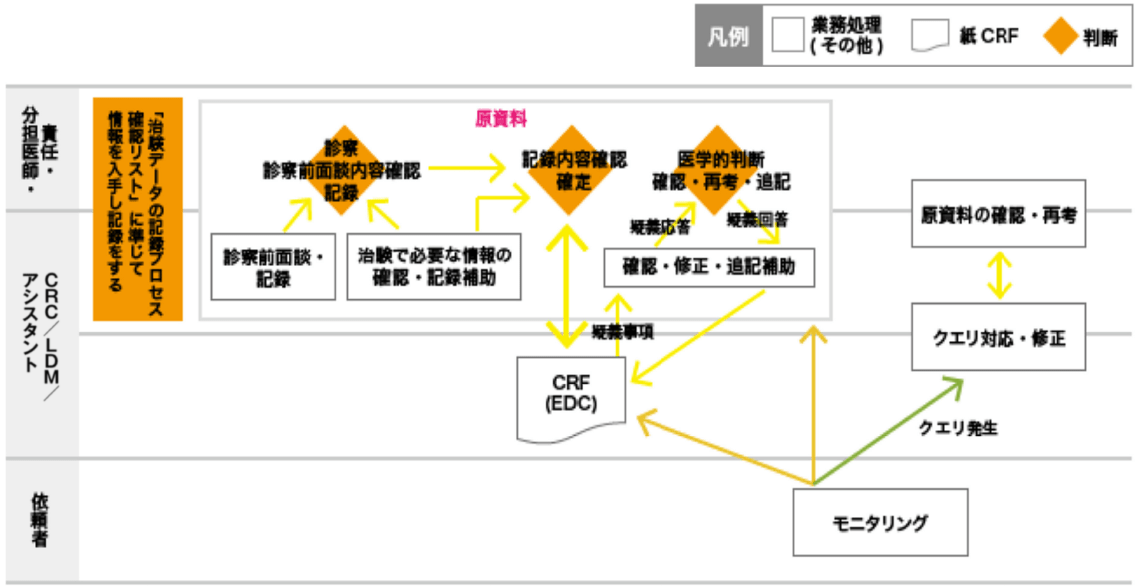
原資料は、「治験データの記録プロセス確認リスト」に準じて、情報を入手し記録をする。
被験者対応時の記憶などに頼らないCRF作成の為に、被験者対応者・原資料作成補助(CRC)とCRF作成者(LDM/アシスタント)を区別する。
1)CRCは被験者対応時、原資料作成を補助する。
2)LDM/アシスタントは原資料からの転記が可能な部分についてCRFを作成する。疑義事項がある場合は、CRCへ疑義照会を行う。
3)CRCは疑義事項の内容に関して対応し、必要に応じて責任(分担)医師へ確認する。LDM/アシスタントは再度原資料を確認しCRFを作成する。
4)クエリの内容を確認し、原資料から読み取れる軽微なものはLDM/アシスタントでクエリ対応する。医学的判断を伴うものについては、3)を繰り返す。
5)試験毎に定められた報告期限までにCRFを完成させる。
資料⑪:逸脱発生時の流れ
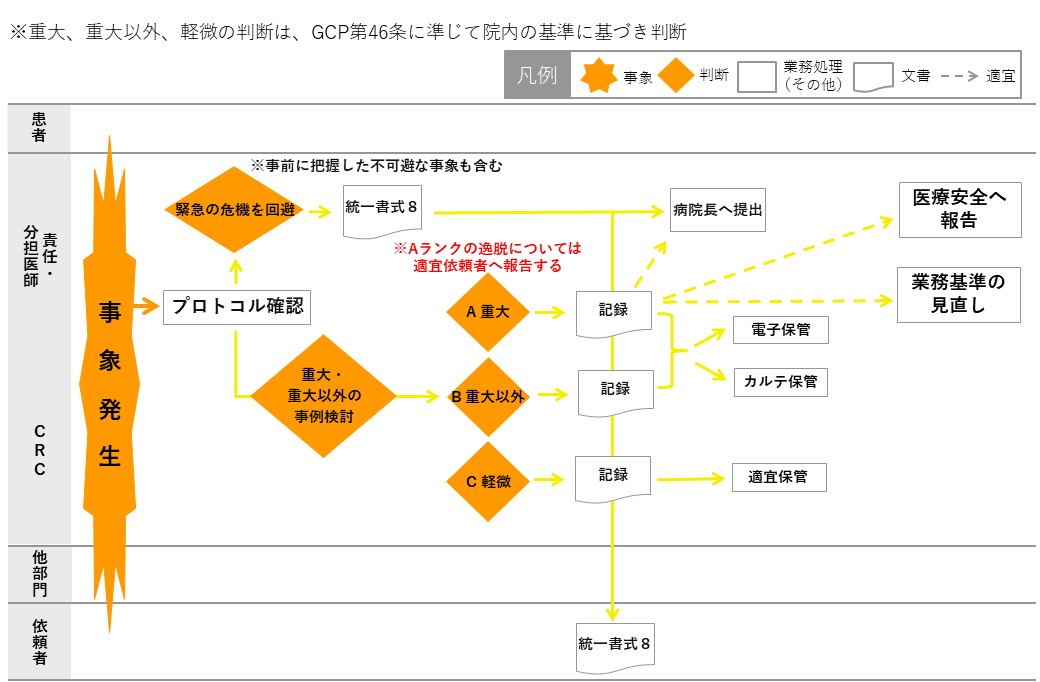
CRCは責任(分担)医師及び室長に報告し、逸脱について記録する。
緊急回避の逸脱の場合には、速やかに報告書(書式8)を作成し、病院長・依頼者へ提出する。
再発防止のために、カンファレンス内で報告・検討を行う。
資料⑫:治験中止・終了時の流れ
※GCP第24条、40条、49条に添って実施
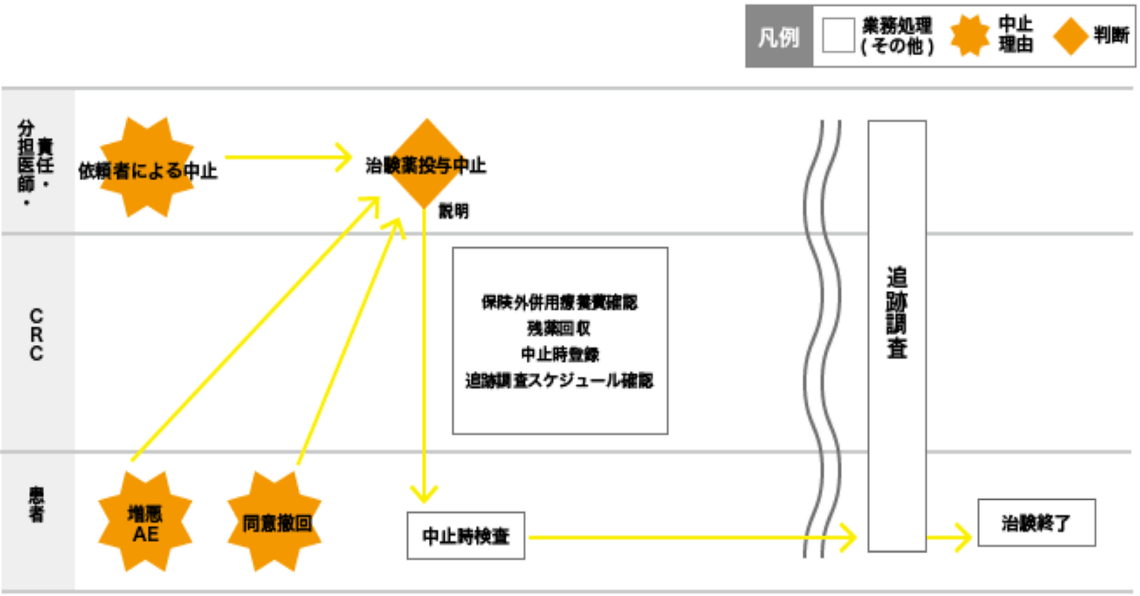
中止や試験終了について被験者への十分な説明を行う。中止・終了後も安心して治療を受けられるように配慮する。さらに、プロトコルで定められた中止時検査や発現中の有害事象の追跡調査などを確認し、必要時支援する。
資料⑬:必須文書の保管について
※GCP第26条、34条、41条、56条に添って実施
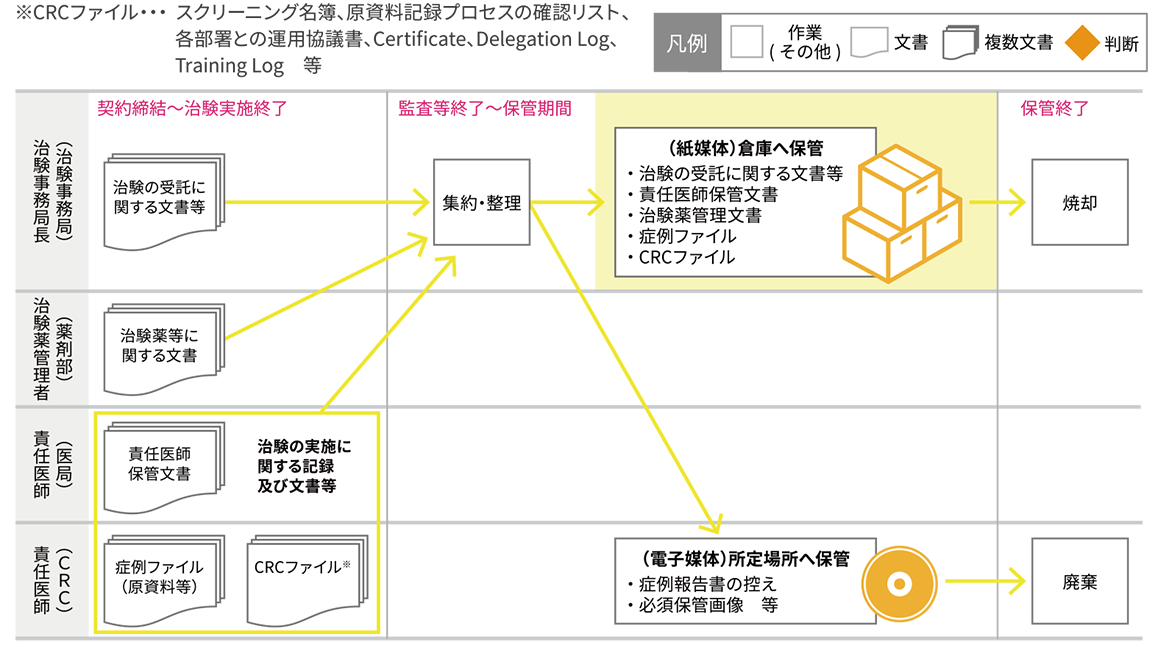
必須文書(治験の受託に関する文書等、責任医師保管文書、治験薬管理文書、症例ファイル、CRCファイル等)は、規定されている期間は当院(委託保管倉庫)で保管・管理する。
※保管が必要なCD-R(症例報告書等)、マスキングが不可能な画像等は、センター内の所定の場所へ保管する。